Medical Education
Medical Education David Gomez Beltran
David Gomez BeltranDavid Villar’s Updates
Elimination of drug from the body

What do mean by elimination of drugs from the body? Elimination is one pharmacokinetic parameter that follows absorption and distribution of drugs following their administration. Elimination is the combination of biotransformation and excretion.
As it names implies, the body is always acting to eliminate foreign substantes when they enter. So whenever we administer a drug, it will do whatever is supposed to do as long as therapeutic levels are maintained, but there are organs like the liver and kidney that will be in charge of the elimination process. So, as you can imagine, from a practical standpoint, that will result in the action being terminated. So, if we know what concentrations are required to attain their therapeutic action, we can estimate how much and how frequent we need to keep administering the drug.
.jpg?Expires=1764966020&Signature=a664WV73gp7rsBcYeBNDqQhiLRIorT96UFtsCJEdIHFrMctKr~tOJl2NWxMBfY~8~zOnD~kb~t1tUGK-YNEDCNBSy95KoQNtForiaMgYz0as7~Aqzmix1F-hqRvlTELQSHnB8o~j3Fgj-X6MFS7se4NyGOicV8kv75gmlmJFEwkCjim1xZSstk8s-oEqFT8f1~y9HAJOv6CIfi1sb1676QboAJaJheAg~0G2A~yec86oRLqJ95f2q6uIXJXuldSGrZMAPcrqqcb-qrNLWYFwyt40AX5leaz3XAMb~m0e3zmPkGplU6mmzZ9Dv~xSoumivokM2artCcUs0xBWrUo~nA__&Key-Pair-Id=APKAJELYXGUCCDL7FUQA)
As already mentioned, there are two steps involved in elimination. The first one is biotransformation ("metabolism" if it is not a xenobiotic), in which enzymes convert the parent molecule into a different one. If we look at other texts, there are many drgus, especially very lipohilic ones that need to be metabolically converted to molecules that are more easily excreted out of the body. Most of those metabolizing enzymes are in the liver and many drug interactions take place because they either inhibit or induce some of those metabolizing enzymes. In the following figure, there is an enzyme that is breaking up a substante into two smaller fractions that we usually call metabolites.
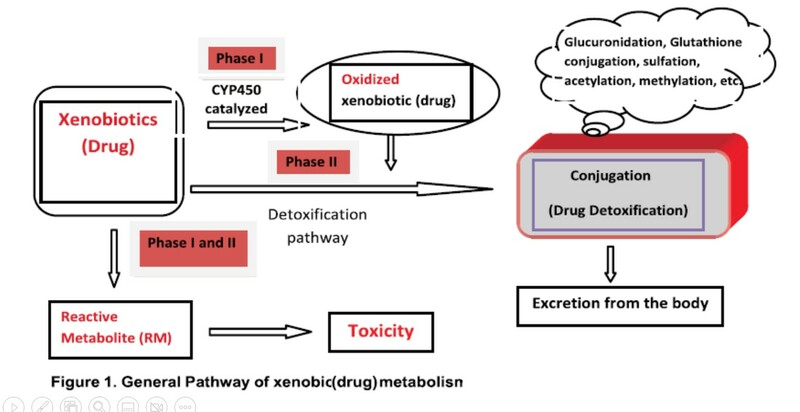
In the next diagram, we can see that xenobiotics may go through two steps, called phase I and II of metabolism. In phase I the drug is converted into a different molecules. In phase II, the drug is combined with body substances (glucuronide, sulfate, etc.) that increases water solubility and can more easily be excreted from the body. Notice that metabolism sometimes results in drugs being converted to more reactive metabolites that can be toxic.
With regards to excretion, the most common route is through the kidneys and out of the body with urine. Obvisouly, the drug needs to be reasonably water soluble to remain in the fluid that becomes the urine. Patients with an impaired kideny function usually have a reduced abilitiy to eliminate hydrophilic drugs, and in order to avoid excessively high concentrations in these patients, we usually need to reduce their dosages or give them less frequently. There are some drugs that enter the bile duct and are excreted in the feces.
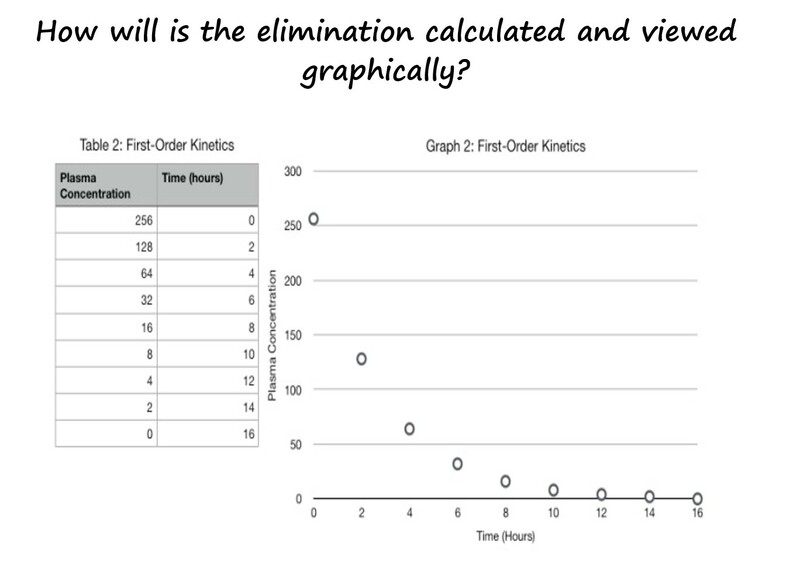
How is the elimination calculated and viewed graphically?
Most drugs are eliminated through what we call 1st order kinetics. So, if were were to collect blood samples at different times following the administration of a drug, in the next graph by IV injection, and measure the concentrations in plasma, we would see a curve that declines overtime.
Because it is difficult to make calculations unless we have a straight line, to determine pharmacokinetic parameters like the T1/2 (half-life), what is usually done is to plot the concentrations in a semilogarithmic scale, and as can be seen when the Y axis is changes into a log scale, the curve now turns into 2 straight lines which correspond to 2 different phases: an intital phase during which the plasma concentration steeply declines (distribution phase) and a second linear phase which is less steep and corresponds to the elimination phase.
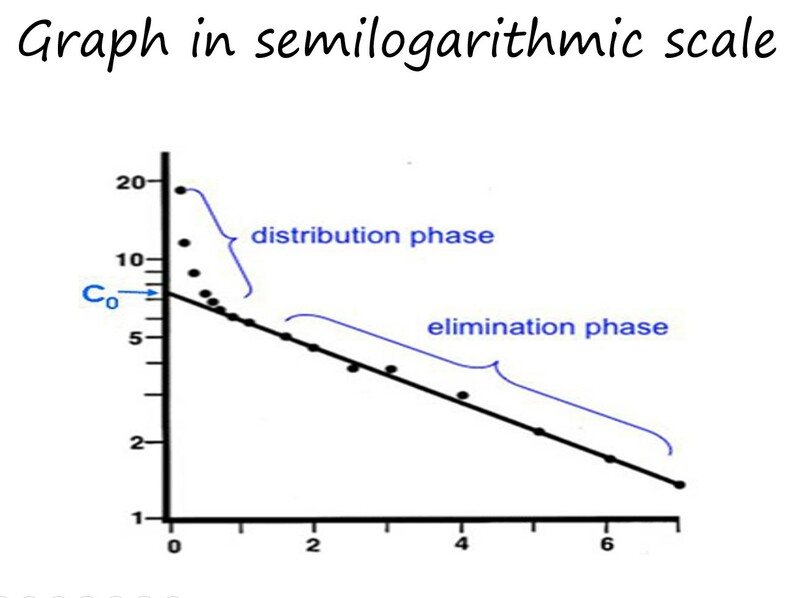
What we see is that after the injetion, the drug will be mainly present in the blood compartment, but it rapidly disributes to the interstitial spaces which is 4 times bigger that the plasma compartment. So, basically the drug is leaving the blood compartment and going into tissues. This early phase usually takes place within minutes for most drugs.
Now, once the drug has distributed into tissues and the concentration reached an equilibrium with those in plasma, the slower decline of the plasma concentrations is associated exclusively with the elimination. Therefore, this phase has two parameters associated with the process and are the constant of elimination (Kel) and the half-life (T1/2). Both can be calculated from the graph and as we will see in another video, they are important in calculating the dosage of maintenance. Kel is the fraction of the drug eliminated in a unit of time. T1/2 is the time is takes for a certain concentration to decline by half.
Another important parameter that needs to be calculated from this graph is the volume of distribution (Vd). If we know the dose given to the animal, we can divide it by the concentration that results from extrapolating the terminal slope to time 0 (Co in the graph). That will give us the Vd.
Vd = dosage/Co
As we will see in the next video, with the Vd and the Kel, we can calculated the maintenance dosage.
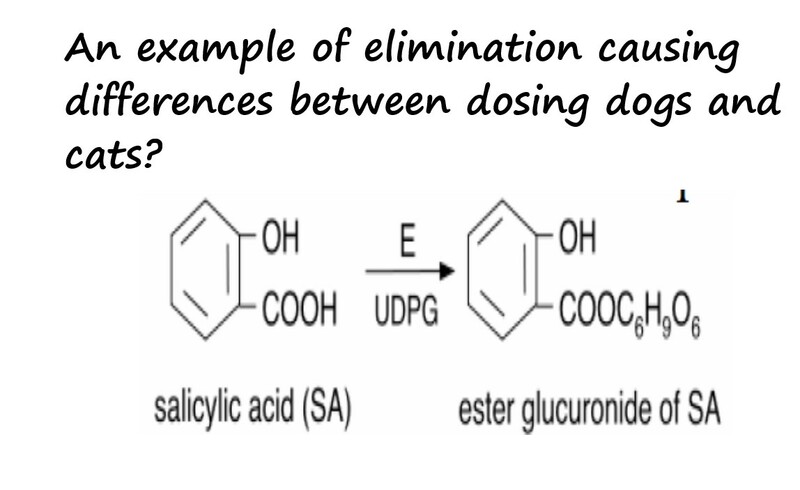
Finally, let´s look at the example below of elimination causing differences between dosing a dogs or a cat. Aspirin is acetyl-salicylic acid. The major elimination pathway involves binding to glucuronic acid to form an ester conjugate that is more soluble and can be easily excreted though the kidneys. Cats lack the enzyme glucuronidase and therfore take longer to eliminate aspirin. This explains why the typical dosage of 10 mg/kg every 8-12 hours in dogsshould be spaced to every other day in cats. This also explains why NSAIDs are more toxic to cats than dogs.